
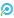
迈基诺全外显子测序DiaWESTM助力发现UPD致LRBA基因纯合变异致病的案例报道
单亲二体(uniparental disomy, UPD),指两条同源染色体均遗传自一个亲本。大部分染色体上的UPD并不会引起相关的临床表型,但是,当UPD存在于6、7、11、14、15,及20号染色体时,由于基因组印记的存在,将会导致异常临床表型的出现。此外,UPD也会导致常染色体上的隐性遗传基因的致病性位点的纯合变异,从而引起致病的发生[1]。
由天津市儿童医院陈森教授团队与北京迈基诺基因科技股份有限公司合作,发现1例罕见父系UPD引起的LRBA基因纯合子变异的病例,该病例是全球第二例UPD致LRBA基因纯合变异致病的报道。这一发现将有助于进一步丰富该基因的突变谱。研究结果于2024年发表在Frontiers in Immunology杂志上,影响因子7.3(见图1)。

图1: 1例罕见父系UPD引起的LRBA基因纯合子变异的病例报道
研究背景
脂多糖反应性米色样锚蛋白(LRBA)缺乏症,是一种由LRBA基因纯合突变引起的原发性免疫缺陷病(PID)。该基因位于4q31.3,编码含有2851个氨基酸的LRBA蛋白,由多个具有重要结构和功能的结构域组成。其中以BEACH结构域的特征最为明显,该结构域长280个氨基酸,在几种蛋白质中都是保守的。LRBA蛋白可在多种细胞类型中表达,其中免疫细胞中的表达量最高,因此,在免疫相关分子的囊泡转运机制中发挥着重要作用[2]。然而迄今为止,与免疫系统相关的LRBA蛋白的生物学作用尚不十分清楚。
LRBA的基因变异在临床上非常罕见,全世界仅报道了两百多例[3]。最初,这种疾病被归类为常见变异性免疫缺陷病,但在不同的 LRBA 患者群被描述后,其表型谱也随之扩大[4,5],将其确定为具有广泛可变表现的临床综合征。慢性腹泻、自身免疫紊乱、器官肥大、频繁的反复感染、低丙种球蛋白血症、慢性肺部表现和生长迟缓,是 LRBA 缺乏症的一些特征。
材料和方法
2.1患者的临床表现
一名两岁男童因发烧和咳嗽 8 天入院。他的出生史并无异常,生长发育与年龄相符,无慢性腹泻病史;既往病史:患儿5个月大时发现中性粒细胞减少症,曾因反复呼吸道感染接受过抗生素治疗;家族史:父母没有血缘关系。
入院前20天,患者出现发热,血常规检查显示中性粒细胞减少和血小板减少(血小板:50×10^9/L)。双肺可闻及啰音和哮鸣音。胸部和腹部 CT:双肺散在炎性合并症;双侧腋窝多发淋巴结肿大,脾脏肿大,腹部和腹膜后多发淋巴结肿大。骨髓穿刺:骨髓细胞增生活跃,粒细胞增生明显,部分粒细胞胞浆颗粒增多。病原体检测:肺炎支原体IgM阳性。
住院期间,医生使用抗生素控制感染。三天后,患者体温恢复正常,但血小板计数降至最低点,仅为4×10^9/L,并伴有皮肤瘀斑。给予1克/千克剂量的静脉免疫球蛋白(IVIG)后,血小板计数增至74×10^9/L。然而,轻度贫血和中性粒细胞减少症依然存在。
2.2 DiaWESTM全外显子组和Sanger 测序
从外周血中提取基因组DNA进行全外显子组测序,由北京迈基诺基因科技股份有限公司完成。测序完成后进行数据注释筛选,对筛选到的可疑致病性变异进行Sanger验证。
结果
DiaWESTM全外显子组测序结果提示,受检人LRBA基因22号外显子中存在一个纯合的无义变异c.2584C>T (p.Gln862Ter) (NM_006726.4)。该变异在HGMD/ClinVar数据库中未见报道。根据 ACMG 指南,初步确定该变异为可能性致病(PVS1+PM2_supporting)。家系Sanger测序结果提示,受检人父亲是该变异的杂合子(无临床表型),母亲不携带该变异(图2)。
图2:通过 Sanger 验证了c.2584C>T(p.Gln862Ter)
UPDio软件分析结果提示,包括LRBA基因在内的4号染色体上存在杂合性缺失(LOH)区域;然而,CNVkit软件分析结果提示,4号染色体上没有缺失区域(图3)。综上推断,LRBA基因的c.2584C>T (p.Gln862Ter) 的纯合状态是由UPD(4)引起的。
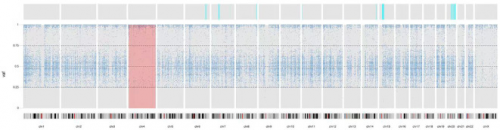
图3: UPD的结果。
4号染色体上的同源变异率大于 90%。VAF,变异等位基因频率。
讨论与小结
Lopez-Herrera等人在2012年首次报道了LRBA基因的功能缺失可导致一种罕见的常染色体隐性先天性免疫失调症,他们的研究揭示了LRBA基因致病变异与免疫缺陷和自身免疫综合征发生之间的相关性。随着基因检测技术的不断进步和临床医学领域对该疾病认识的加深,截至2021年,全球已报告约212例LRBA缺乏症患者。该疾病患者的临床表型变化很大,即使在同一家族中,基因型与表型之间的关系也不完全清楚,而且还可能受到表观遗传或环境因素的影响。
本例患者还伴有肺炎支原体感染,肺炎支原体是导致变异性免疫缺陷(CVID)患者肺部感染的常见病原体。泛发性溶血性贫血是其很少见的症状。在本病例中,患者从5个月大开始出现慢性中性粒细胞减少症和反复呼吸道感染,最终导致全血细胞减少而住院治疗。尽管对感染进行了有效治疗,但血细胞谱无明显改善。骨髓检查排除了原发性血液病和先天性骨髓衰竭综合征,表明血液病症状也是LRBA缺乏症的临床表现。虽然文献报道LRBA缺乏症患者可出现中性粒细胞和血小板减少症,但出现全血细胞减少症的病例并不多见。本例患者的发现丰富了LRBA缺乏症的表型谱。
4号染色体没有明显的疾病相关印记基因,大多数UPD(4)的表型主要是由隐性基因的表现引起的。由UPD引起的先天性免疫错误(IEI)的报告极为罕见。2018年,西班牙研究人员报告了首例由mUPD引起的新型同基因LRBA基因变异病例,揭示了这种罕见的基因变异类型。该病例是全球第二例由UPD引起的新型纯合LRBA基因变异的报道,它是pUPD的较罕见形式。在中国,关于LRBA基因变异的报道很少,也缺乏系统的研究。该患者的全外显子组测序结果显示,LRBA基因第22号外显子上存在一个c.2584C>T (p.Gln862Ter) 的纯合变异,预计该变异会导致一个过早的终止密码子,导致LRBA蛋白的C端(包括BEACH结构域)丢失2000多个氨基酸,从而导致LRBA蛋白的功能缺失引起疾病的发生。
LRBA缺乏症是近十年来新发现的一种遗传性免疫缺陷病。患者人数相对较少,新的基因变异类型仍在不断被发现。随着基因检测技术的不断进步,预计将有更多患者得到诊断和治疗。
迈基诺DiaWESTM全外显子组基因检测,不仅可以检测点突变、微小插入或缺失等,此外,还会针对UPD进行分析,在这个病例中,准确的检出了单亲二体的情况,以此完成对患者的精准诊断,同时也为LRBA基因变异数据库提供了新的突变体和临床数据。
参考文献
Yamazawa K, Ogata T, Ferguson-Smith AC. Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet. 2010 Aug 15;154C(3):329-34..
Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas C, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90:986–1001.
Kardelen AD, Kara M, Güller D, Ozturan EK, Abalı ZY, Ceylaner S, Kıykım A, Cantez S, Torun SH, Poyrazoglu S, Bas F, Darendelıler F. LRBA deficiency: a rare cause of type 1 diabetes, colitis, and severe immunodeficiency. Hormones (Athens). 2021 Jun;20(2):389-394.
Azizi G, Abolhassani H, Mahdaviani SA, Chavoshzadeh Z, Eshghi P, Yazdani R, Kiaee F, Shaghaghi M, Mohammadi J, Rezaei N, Hammarström L, Aghamohammadi A (2017) Clisnical, immunologic, molecular analyses and outcomes of Iranian patients with LRBA deficiency: a longitudinal study. Pediatr Allergy Immunol 28:478–4842.
Gámez-Díaz L, Seidel M. Different Apples, Same Tree: Visualizing Current Biological and Clinical Insights into CTLA-4 Insufficiency and LRBA and DEF6 Deficiencies. Front Pediatr. 2021;9:662645.
迈基诺全外显子组测序DiaWESTM:
1. 精准检测:自主专利GenCap®高保真性DNA双链探针,高效稳定的进行变异检测,结合十年积累的特殊算法可以更好分析CNV。
2. 智能设计:BaitDesigner™ 探针设计系统实现探针智能化设计,针对特殊基因实现探针密度和覆盖度的灵活调整,,以及精准分析CNV和上万个非编码区变异,同时全长覆盖线粒体环基因。
3. 分析全面:覆盖重点关注的热点区域、基因外显子以及内含子区域,同时包括可发现单个外显子水平的CNV、地贫缺失型、希特林蛋白基因3kb插入、SMN1基因7号外显子缺失、线粒体环基因变异、染色体大范围的单亲二体和杂合性缺失、动态突变等。
4. 精准注释:依托于诺云平台™ 已知公共数据库+迈基诺自建34万余人的中国人群数据库+商业数据库,让基因测序数据解读更加精准,节约90%以上的人工解读工作。
环球健康报



 杜汶泽婚姻羡煞旁人 称人生找到好太太
杜汶泽婚姻羡煞旁人 称人生找到好太太 维多利亚减肥秘密 向世界传达女性美
维多利亚减肥秘密 向世界传达女性美 气质女王刘诗诗 瘦人体质不发胖
气质女王刘诗诗 瘦人体质不发胖 萧亚轩"瘦美人" 被柯震东疯狂示爱
萧亚轩"瘦美人" 被柯震东疯狂示爱